| Structure info | |
|---|---|
| Layer group | p2_122 |
| Layer group number | 20 |
| Structure origin | Lyngby22_LDP |
| Stability | |
|---|---|
| Energy above convex hull [eV/atom] | 0.003 |
| Heat of formation [eV/atom] | -0.234 |
| Dynamically stable | No |
| Basic properties | |
|---|---|
| Magnetic | No |
| Band gap (PBE) [eV] | 1.112 |
| Symmetries | |
|---|---|
| 2D Bravais type | Rectangular (op) |
| Layer group number | 20 |
| Layer group | p2_122 |
| Space group number (bulk in AA-stacking) | 17 |
| Space group (bulk in AA-stacking) | P222_1 |
| Point group | 222 |
| Inversion symmetry | No |
| Structure data | |
|---|---|
| Formula | Ag2Se4Cl2 |
| Stoichiometry | ABC2 |
| Number of atoms | 8 |
| Unit cell area [Å2] | 42.886 |
| Thickness [Å] | 3.391 |
| Ag2Cl2Se4 (2AgClSe2-2) | |
|---|---|
| Heat of formation [eV/atom] | -0.23 |
| Energy above convex hull [eV/atom] | 0.00 |
| Monolayers from C2DB | |
|---|---|
| Ag3Cl3 (3AgCl-1) | -0.44 eV/atom |
| Ag2Cl2 (2AgCl-1) | -0.44 eV/atom |
| Ag6Cl6 (6AgCl-1) | -0.43 eV/atom |
| Ag4Cl4 (4AgCl-1) | -0.43 eV/atom |
| Ag3Cl3 (3AgCl-2) | -0.43 eV/atom |
| Ag6Cl6 (6AgCl-2) | -0.43 eV/atom |
| Ag12Cl12 (12AgCl-1) | -0.43 eV/atom |
| Ag2Cl2 (2AgCl-2) | -0.42 eV/atom |
| Ag6Cl6 (6AgCl-3) | -0.42 eV/atom |
| Ag6Cl6 (6AgCl-4) | -0.42 eV/atom |
| Ag3Cl3 (3AgCl-3) | -0.42 eV/atom |
| Ag4Cl4 (4AgCl-2) | -0.42 eV/atom |
| Ag4Cl4 (4AgCl-3) | -0.41 eV/atom |
| Ag2Cl2 (2AgCl-3) | -0.41 eV/atom |
| Ag8Cl8 (8AgCl-1) | -0.41 eV/atom |
| Ag2Cl2 (2AgCl-4) | -0.41 eV/atom |
| Ag16Cl16 (16AgCl-1) | -0.41 eV/atom |
| Ag8Cl8 (8AgCl-2) | -0.40 eV/atom |
| AgCl (1AgCl-1) | -0.40 eV/atom |
| Ag3Cl3 (3AgCl-4) | -0.40 eV/atom |
| Ag2Cl2 (2AgCl-5) | -0.40 eV/atom |
| Ag2Cl2 (2AgCl-6) | -0.39 eV/atom |
| Ag2Cl2 (2AgCl-7) | -0.39 eV/atom |
| Ag4Cl4 (4AgCl-4) | -0.38 eV/atom |
| Ag2Cl4 (2AgCl2-1) | -0.36 eV/atom |
| Ag4Cl8 (4AgCl2-1) | -0.36 eV/atom |
| AgCl2 (1AgCl2-1) | -0.29 eV/atom |
| Ag4Cl8 (4AgCl2-2) | -0.28 eV/atom |
| Ag4Cl4Se4 (4AgClSe-1) | -0.24 eV/atom |
| Ag2Cl2Se4 (2AgClSe2-1) | -0.24 eV/atom |
| Ag2Cl2Se4, (2AgClSe2-2) | -0.23 eV/atom |
| AgCl2 (1AgCl2-2) | -0.20 eV/atom |
| AgCl3 (1AgCl3-1) | -0.18 eV/atom |
| AgCl2 (1AgCl2-3) | -0.17 eV/atom |
| Ag2Cl6 (2AgCl3-1) | -0.15 eV/atom |
| Ag2Cl2Se2 (2AgClSe-1) | -0.11 eV/atom |
| Se2Ag4 (2SeAg2-1) | -0.09 eV/atom |
| Se8Ag16 (8SeAg2-1) | -0.07 eV/atom |
| Ag2Cl6 (2AgCl3-2) | -0.04 eV/atom |
| AgClSe (1AgClSe-1) | -0.04 eV/atom |
| Ag4Se4 (4AgSe-1) | -0.02 eV/atom |
| Ag2Se2 (2AgSe-1) | 0.02 eV/atom |
| Ag8Se12 (4Ag2Se3-1) | 0.03 eV/atom |
| Ag2Se4 (2AgSe2-1) | 0.05 eV/atom |
| Ag2Se2 (2AgSe-2) | 0.05 eV/atom |
| Ag2Se2 (2AgSe-3) | 0.06 eV/atom |
| AgSe2 (1AgSe2-1) | 0.11 eV/atom |
| SeAg2 (1SeAg2-1) | 0.15 eV/atom |
| Ag2Se2 (2AgSe-4) | 0.15 eV/atom |
| Ag2Se2 (2AgSe-5) | 0.18 eV/atom |
| Ag2Se4 (2AgSe2-2) | 0.19 eV/atom |
| Se2 (2Se-1) | 0.21 eV/atom |
| AgSe2 (1AgSe2-2) | 0.22 eV/atom |
| Ag2Se2 (2AgSe-6) | 0.27 eV/atom |
| Se2 (2Se-2) | 0.29 eV/atom |
| Ag2 (2Ag-1) | 0.31 eV/atom |
| AgSe2 (1AgSe2-3) | 0.32 eV/atom |
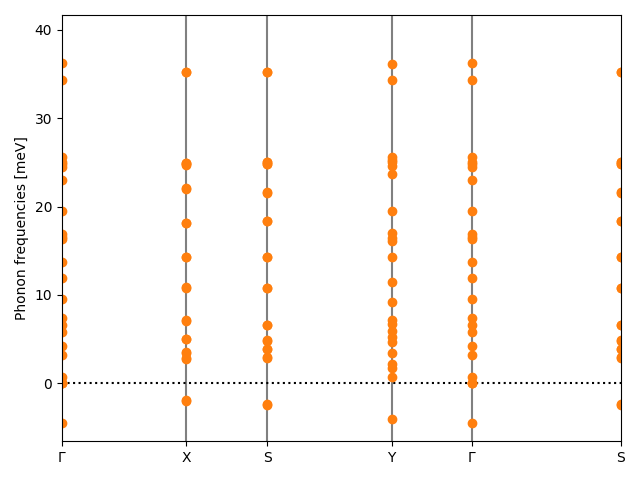
| Minimum eigenvalue of Hessian [eV/Ų] | -0.20 |
| Cij (N/m) | xx | yy | xy |
| xx | 10.33 | 1.68 | -0.01 |
| yy | 3.11 | 7.82 | -0.01 |
| xy | 0.00 | 0.00 | 8.07 |
| Stiffness tensor eigenvalues | |
| Eigenvalue 0 | 6.47 N/m |
| Eigenvalue 1 | 8.07 N/m |
| Eigenvalue 2 | 11.68 N/m |
| Key values [eV] | |
|---|---|
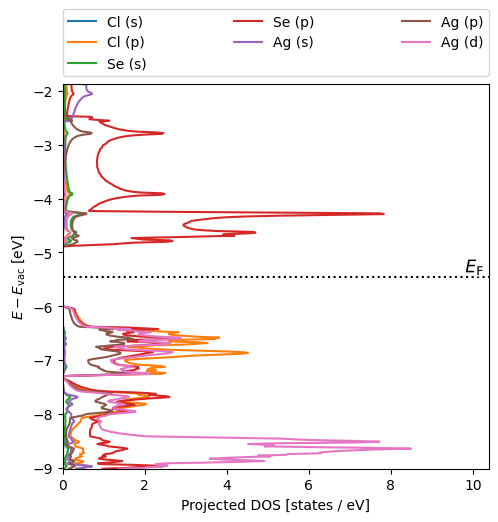
| Band gap (PBE) | 1.112 |
| Direct band gap (PBE) | 1.164 |
| Valence band maximum wrt. vacuum (PBE) | -6.005 |
| Conduction band minimum wrt. vacuum (PBE) | -4.894 |
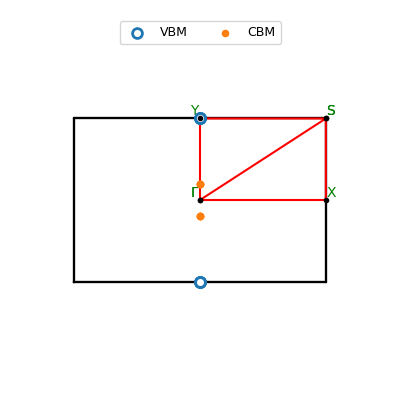
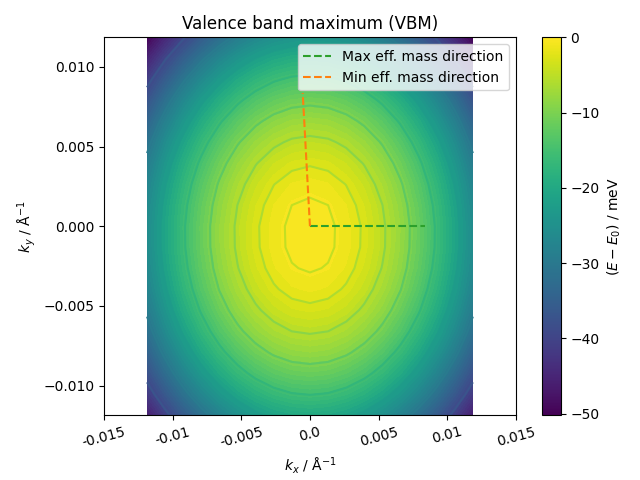
| Property (VBM) | Value |
|---|---|
| Min eff. mass | 0.88 m0 |
| Max eff. mass | 1.09 m0 |
| DOS eff. mass | 0.97 m0 |
| Crystal coordinates | [0.000, -0.496] |
| Warping parameter | -0.002 |
| Barrier height | > 19.9 meV |
| Distance to barrier | > 0.0119 Å-1 |
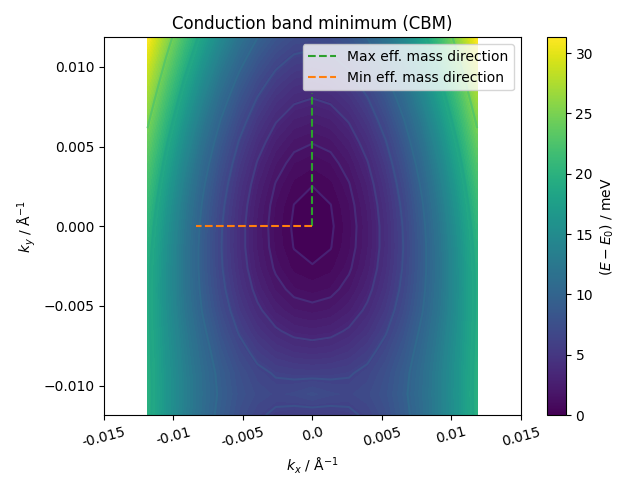
| Property (CBM) | Value |
|---|---|
| Min eff. mass | 0.99 m0 |
| Max eff. mass | 2.35 m0 |
| DOS eff. mass | 1.52 m0 |
| Crystal coordinates | [0.000, 0.085] |
| Warping parameter | 0.005 |
| Barrier height | > 6.7 meV |
| Distance to barrier | > 0.0125 Å-1 |
| Atom No. | Chemical symbol | Charges [|e|] |
|---|---|---|
| 0 | Cl | -0.57 |
| 1 | Cl | -0.57 |
| 2 | Se | 0.08 |
| 3 | Se | 0.08 |
| 4 | Se | 0.07 |
| 5 | Se | 0.07 |
| 6 | Ag | 0.42 |
| 7 | Ag | 0.42 |
| Miscellaneous details | |
|---|---|
| Unique ID | 2AgClSe2-2 |
| Number of atoms | 8 |
| Number of species | 3 |
| Formula | Ag2Se4Cl2 |
| Reduced formula | AgSe2Cl |
| Stoichiometry | ABC2 |
| Unit cell area [Å2] | 42.886 |
| Original file-system folder | /home/niflheim2/cmr/C2DB-ASR/tree_LDP/ABC2/AgClSe2/Ag2Cl2Se4-d48005e93f58 |
| Old uid | Ag2Cl2Se4-0bc7db0db50c |
| Space group (bulk in AA-stacking) | P222_1 |
| Space group number (bulk in AA-stacking) | 17 |
| Point group | 222 |
| Inversion symmetry | No |
| Layer group number | 20 |
| Layer group | p2_122 |
| 2D Bravais type | Rectangular (op) |
| Thickness [Å] | 3.391 |
| Structure origin | Lyngby22_LDP |
| Miscellaneous details | |
|---|---|
| Band gap (PBE) [eV] | 1.112 |
| Direct band gap (PBE) [eV] | 1.164 |
| gap_dir_nosoc | 1.214 |
| Vacuum level [eV] | 2.522 |
| Fermi level wrt. vacuum (PBE) [eV] | -5.450 |
| Valence band maximum wrt. vacuum (PBE) [eV] | -6.005 |
| Conduction band minimum wrt. vacuum (PBE) [eV] | -4.894 |
| minhessianeig | -0.195 |
| Dynamically stable | No |
| Energy [eV] | -25.054 |
| Magnetic | No |
| Total magnetic moment [μB] | 0.000 |
| Spin axis | z |
| Magnetic anisotropy energy, xz [meV/unit cell] | 0.000 |
| Magnetic anisotropy energy, yz [meV/unit cell] | 0.000 |
| Energy above convex hull [eV/atom] | 0.003 |
| Heat of formation [eV/atom] | -0.234 |
